The rapid advancement of the cell and gene therapy industry sometimes precedes regulatory guidance. That makes it complex to navigate international regulations for cellular starting material.
When you procure cellular starting material or distribute a cell or gene therapy product across international borders, you must consider the country or regional requirements at play and where they diverge.
Allogeneic cell therapy sponsors face common challenges when treating a global patient population with therapies using healthy adult donor starting material. Challenges include:
- Differences in donor eligibility requirements between countries
- The impact of donor compensation on global distribution
- Differences in allogeneic source material collection requirements
- Lack of clarity around when good manufacturing practices (GMP) are applicable in the cell collection process
Table 1. Example of international regulatory differences in source material collection
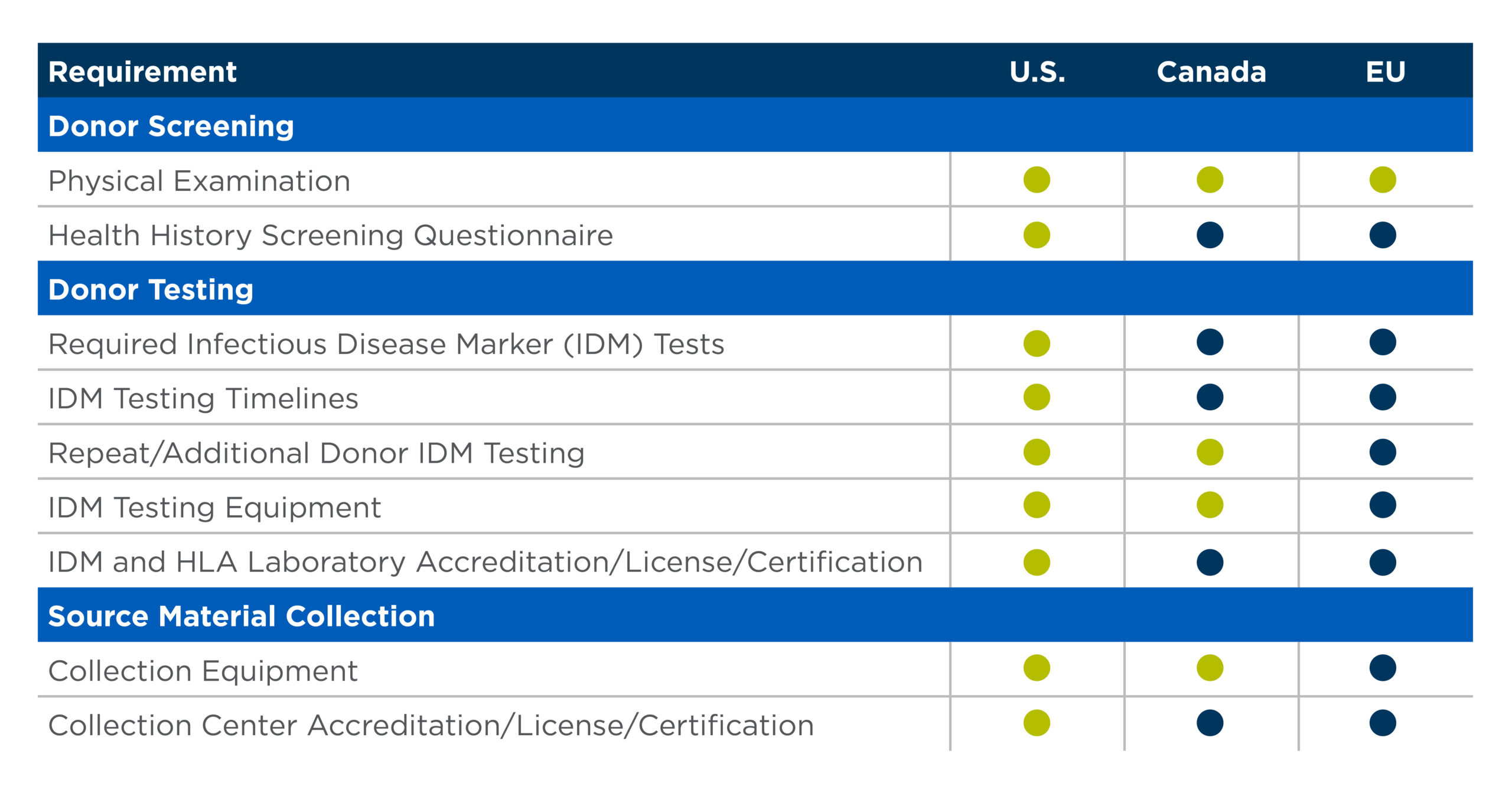
As Table 1 demonstrates, collecting starting material in one region is not a plug and play for a different region.
The table uses United States requirements as a starting point. It illustrates at a high level the areas where regulatory differences exist between the U.S., Canada and the European Union.
It is critical to understand what those differences are and build them into your cell therapy development processes.
Differences in donor eligibility requirements between countries
Eligibility requirements generally fall into two categories: donor screening and donor testing. Donor screening typically includes activities such as a donor physical examination, administration of a donor health history questionnaire and review of other relevant medical records. Donor physical examination requirements tend to be similar across regions but donor health history assessments through questionnaires can have significant variation.
The donor testing required may also vary between regions.
For instance, in the U.S., donors are required to be tested for infectious diseases outlined in 21 CFR 1271, which includes hepatitis B and C, HIV, syphilis, West Nile virus and CMV. In Europe, particularly depending on any Member States of interest, additional infectious disease testing requirements may be added, such as hepatitis A and E, parvovirus B19 and Toxoplasma gondii.
Donor testing timelines and other testing requirements can also vary.
For example, in the U.S. the typical window for taking a donor sample for IDM testing is seven days pre-collection to seven days post-collection. That is a window of 15 days. In Canada, that timeline is within 30 days before collection. In the EU, samples must typically be taken on the day of collection or within seven days post collection.
Testing requirements for kits and laboratories is another good example.
In the U.S., appropriate FDA-licensed, approved or cleared donor testing kits must be used in accordance with the manufacturer’s instructions in a laboratory that is certified under the Clinical Laboratory Improvement Amendments (CLIA) or has met equivalent requirements as determined by the Centers for Medicare and Medicaid Services (CMS). In Europe, testing must be performed on CE marked test kits whenever possible and may require additional laboratory certifications.
Impact of donor compensation on global distribution
You must pay close attention to whether a country in which you will distribute your therapy allows donor compensation.
Compensation means you pay a donor in response to their ability or willingness to donate cells.
That is different from reimbursement, which makes the donation accessible for the donor. Paying for a donor to travel for donation is an example of reimbursement.
Donor compensation may improve donor follow through to donation or increase the likelihood the donor will return for a repeat donation. However, there are significant cons.
You may not have the same level of freedom in allogeneic cell therapy product distribution if you compensate donors. Key international regions currently either strongly emphasize or require the use of voluntary, unpaid donors for cellular therapies. These include the EU, Canada and Japan.
In addition, current standards from industry organizations and accrediting agencies, such as the Foundation for the Accreditation of Cellular Therapy (FACT) and the World Marrow Donor Association (WMDA), recommend not compensating donors.
Before implementing a donor compensation or expense reimbursement program, consult with your legal counsel to determine if there may be any impacts depending on the global jurisdiction in which you plan to distribute your therapy.
Differences in starting material collection requirements
You may be asking yourself, “How can I assess donors and collect cellular starting material in a way that complies with multiple international regions?”
Unfortunately, there isn’t a standard way of doing this. Regulatory requirements for starting material collection in different countries are still catching up with the advancements in cell therapy.
However, it could help to:
- Take international considerations into account from the beginning as you establish your supplier qualification process and requirements.
- Reach out to colleagues in the country where the cells will be collected to understand that country’s collection practices and regulations.
- Review the requirements in the country where the cells will be collected. Assess if they match the requirements of the country in which the therapy will be distributed.
Lack of clarity around when GMP is applicable in the cell collection process
The question of whether GMPs apply to the cell collection process and starting material processing has been unclear for some time. However, the FDA has recently provided some clarity for the U.S.
During an FDA Office of Tissues and Advanced Therapies (OTAT) Town Hall on Sept. 29, 2022, the FDA said that GMP does not apply to the collection of allogeneic starting material. Specifically, an FDA official said companies only need to adhere to 21 CFR 1271 subpart C and do not need to collect allogeneic starting material under full GMP conditions. The requirement for adherence to GMPs starts at the manufacturing sites.
The FDA does, however, recommend cell therapy companies implement standard procedures for the collection, storage and shipment of material, especially if collections occur at multiple sites.
In the EU, some countries believe GMP standards should apply to the procurement of source material. Others believe that the producer of starting material does not need to be GMP compliant if the collection site is regularly inspected or audited by a regulatory authority. In the U.K. and Canada, there are currently no specific requirements for GMP practices during the collection process.
Given the differences, there is uncertainty in which way the industry will trend. Some sponsors may have already built GMP into their starting material collection processes and commitments to agencies. It remains to be seen if GMP will continue to be an expectation for collection facilities or if it will be laid aside over time.
Resources to assess key regulations for allogeneic cellular source material
Our team at NMDP BioTherapies℠ works with cell and gene therapy sponsors who plan to distribute their allogeneic therapies in multiple countries. We’ve found the following resources helpful as a starting point to assess key regulations in the U.S., Canada, the U.K. and the EU.
This list should not be considered exhaustive, and regulations are subject to change.
For an in-depth discussion on these topics, watch International Differences in Cellular Starting Material Quality and Regulatory Requirements. The one-hour on-demand webinar features perspectives from panelists from the U.S., Canada, U.K. and EU.
Additional resources that may interest you
The CAR-T Cell Handbook offers clinical care applications for health care professionals that treat patients with CAR-T cell therapies. It is a joint venture between the European Society of Blood and Marrow Transplantation (EBMT) and the European Hematology Association (EHA).
This resource reviews current mononuclear cell procurement practices in the Advanced Therapy Medicinal Product (ATMP) community. It offers recommendations on standardized approaches to reduce unnecessary complexity and variation.





